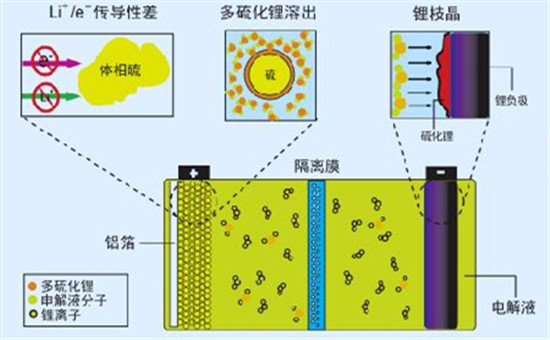
采用插層技術對鋰硫電池的“穿梭”效應和倍率性能進行改性。
理論結合實驗,證實了錫原子插層后的MoO3對多硫化鋰具有更強的吸附性能及更好的導電性能。
相對于傳統的鋰離子電池,鋰硫電池具有理論能量密度高、對環境友好、價格低廉等優勢,在過去的幾年里引起了科研界和商業屆人士的眾多關注。然而,由于其自身的諸多問題,鋰硫電池的商業化進程受到了一定程度上的阻礙。鋰硫電池的主要問題體現在如下兩個方面:1. 在放電過程中形成的多硫化鋰(LiPSs) 容易溶解到電解液中,并穿過半透膜沉積到負極金屬鋰的表面,不僅導致容量的衰減還阻礙了充放電過程的進一步進行;2. 活性物質硫(S8)及放電產物Li2S2-x (x ≤ 1)的絕緣特性,導致電池的倍率性能較差。因此,引入一種理想的載體,不僅對LiPSs具有較強的吸附性,而且具有較高的導電性顯得尤為重要。
具有較強極性的金屬氧化物(MOs)受到了研究者廣泛的關注,但是絕大部分的MOs的導電性較差,不利于鋰硫電池倍率性能的發揮。最近,北京航空航天大學的宮勇吉教授課題組提出了使用插層的方法調控二維材料電化學性質的概念。作者采用二維的三氧化鉬(MoO3)為前驅體,采用插層技術將錫(Sn)原子插入到MoO3的范德瓦爾斯層間,合成的Sn插層MoO3 (Sn-MoO3)的電導率有較大幅度的提升,對鋰硫電池的倍率性能有了明顯的改善。此外,Sn-MoO3與四硫化二鋰(Li2S4)之間的結合能明顯高于MoO3和傳統的石墨烯材料,有效的抑制了LiPSs的“穿梭”效應,極大的提高的鋰硫電池的循環穩定性。該文章發表在國際頂級期刊Advanced Energy Materials上(IF:21.875),其中第一作者為北京航空航天大學的楊偉偉博士和肖杰文本科生,通訊作者為宮勇吉教授和張千帆副教授。
本文的亮點在于首次將插層技術應用到鋰硫電池的研究中,通過插層技術將金屬原子插入到二維層狀MOs的范德瓦爾斯層間,不僅有效的提高了鋰硫電池的倍率性能,還有效的抑制了“穿梭”效應的發生。
在本工作中,研究者首先合成厚度 ~10nm的二維MoO3納米帶,然后通過歧化反應將Sn插入到MoO3的范德瓦爾斯層間。通過顏色變化表明,插入不同量的Sn后,SnxMoO3(0.028≤x≤0.063) 的顏色逐漸從白色(MoO3)變成淺藍,最后變成深藍色。通過TEM測試表明,Sn均勻的插入到了MoO3的范德瓦爾斯層間,而不是負載在材料的表面;從XRD結果可以以得出Sn0.028MoO3, Sn0.050MoO3, Sn0.063MoO3的層間距從0.6954 nm(MoO3) 逐漸增加到0.7076,0.7350, 0.7544 nm;從粉末電導率測試表明電導率從1.31 (MoO3) 增加至2.05 S m-1 (Sn0.063MoO3);UV-vis進一步表明材料的帶隙從3.05 eV (MoO3)減小到 2.23 eV (Sn0.063MoO3)。

圖1. MoO3 納米帶及其插入不同含量Sn的溶液顏色變化圖。a) 純 MoO3, b) Sn0.028MoO3,c) Sn0.050MoO3,d) Sn0.063MoO3。
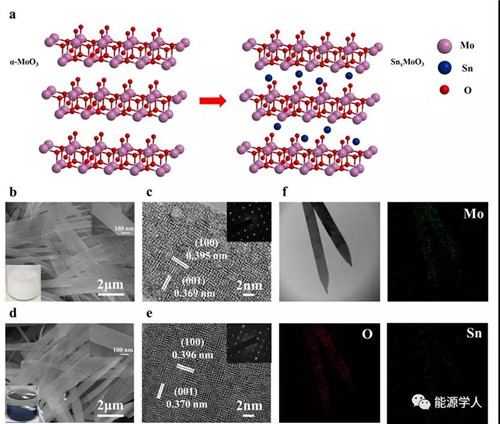
圖2. MoO3和Sn0.063MoO3的合成與表征。a)Sn原子嵌入MoO3納米帶的過程示意圖,b)MoO3納米帶的SEM圖。插圖:以丙酮為溶劑的MoO3溶液顏色圖和MoO3的SEM放大圖,c)MoO3的HRTEM圖。插圖:MoO3的SAED圖,d)Sn0.063MoO3納米帶的SEM圖。插圖:以丙酮為溶劑Sn0.063MoO3顏色圖和Sn0.063MoO3放大的SEM圖,e)Sn0.063MoO3的HRTEM圖。插圖:Sn0.063MoO3的SAED圖,f)原始TEM圖像和Mo、O和Sn的元素分布圖。
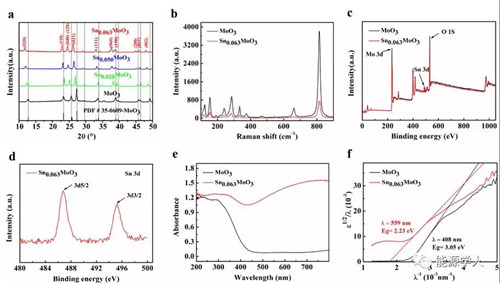
圖3. MoO3和Sn0.063MoO3的表征。a)MoO3和SnxMoO3的XRD測試,b)MoO3和Sn0.063MoO3的拉曼光譜,c)完整的XPS光譜表明MoO3在插層后有明顯的Sn峰,d)Sn的3d5/2和3d3/2的XPS光譜,e)和f)為MoO3和Sn0.063MoO3的UV-vis吸收光譜。
其次,通過DFT進一步計算了MoO3,Sn0.063MoO3與S8,Li2S, Li2S4之間的結合能大小。結果表明,Sn-MoO3與四硫化二鋰(Li2S4)之間的結合能高達3.01 eV, 高于MoO3(2.44 eV)和傳統石墨烯(0.10 eV)與Li2S4之間的結合能,結合能的提升可以有效的抑制了LiPSs的“穿梭”效應,極大的提高鋰硫電池的循環性能。

圖4. DFT理論計算。 a) Li2S4在MoO3和Sn-MoO3表面的優化吸附構型,b)分別計算了石墨烯、MoO3和Sn-MoO3與S8、Li2S4和Li2S的結合強度,具體結合強度(eV)見柱狀圖,c, d) 分別為MoO3和Sn-MoO3的能態密度(DOS),e, f) 分別為Li2S4吸附在MoO3和Sn-MoO3表面的態密度。
最后,作者研究了MoO3-S和Sn0.063MoO3-S電極的電化學性質。Sn0.063MoO3-S電極在1 C條件下的放電容量高達902.6 mAh g–1,而在相同的電流密度下MoO3-S電極放電容量只能達到530.6 mAh g–1,而且從充放電曲線可以看出,Sn0.063MoO3-S電極的極化明顯小于MoO3-S。此外,Sn0.063MoO3-S電極在0.1, 0.2, 0.5, 1, 2, 3和 4 C條件下的放電容量分別為1390.3,1235.3,1032.5,902.9,762.3,638.5和529.7 mAh g-1,而且將電流重新轉變成0.1 C時,Sn0.063MoO3-S電極的放電容量依然可以達到1212.3 mAh g-1,表現出了較好的倍率性能和循環性能。而且,Sn0.063MoO3-S電極在1C充放電條件下循環500次,容量保持率高達79.6%,進一步驗證了其優異的循環性能。
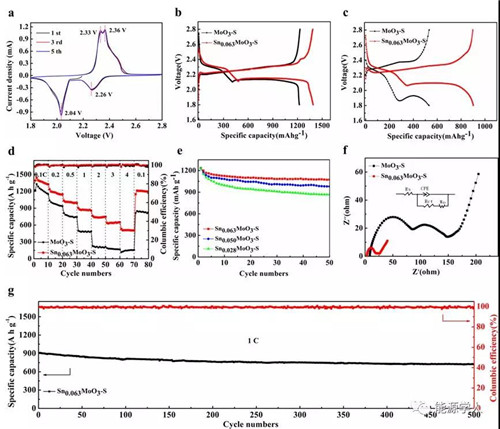
圖5. MoO3-S和Sn0.063MoO3-S電極的電化學性質測試。a) Sn0.063MoO3-S的CV曲線測試,掃描速率為0.05 mV s-1, MoO3-S和Sn0.063MoO3-S的充放電曲線: b) 0.1 C, c) 1 C, d) MoO3-S和Sn0.063MoO3-S的倍率性能測試,e) Sn0.028MoO3-S、Sn0.050MoO3-S、Sn0.063MoO3-S在0.2 C條件下的循環性能,f) 5個循環后,Sn0.063MoO3-S和MoO3-S的EIS測試,g) Sn0.063MoO3-S電極在1C條件下的循環性能。
綜上所述,插層后的Sn0.063MoO3-S電極表現出了優異電化學性能的可能原因如下:(1)插層后Sn0.063MoO3的導電性相對于純相MoO3有了很大程度的改善,有利于鋰硫電池倍率性能的發揮;(2)插層后的Sn0.063MoO3與Li2S4之間具有較強的結合能,有效的抑制了“穿梭”效應的發生,明顯的提高了鋰硫電池的循環壽命。
【材料制備過程】
SnxMoO3粉末的制備: Sn通過Sn(II)的歧化氧化還原反應插入到MoO3范德瓦爾斯層間。首先,將144 mg MoO3納米帶均勻的分散在丙酮溶液中,然后在攪拌的條件下先后加入 90 mg SnCl2和630 mg酒石酸,在80℃的條件下反應3 h,反應結束后用丙酮潤洗3次。最后,將藍色粉末標記為Sn0.063MoO3。同時,將SnCl2和酒石酸的含量分別改變為10、70 mM和30、210 mM,得到的粉末分別記為Sn0.028MoO3和Sn0.050MoO3,其他條件保持一致。
MoO3-S和SnxMoO3-S電極的制備:采用常見的的熔融擴散方法制備了MoO3-S和SnxMoO3-S復合材料。首先,將合成的0.6 g MoO3、Sn0.028MoO3、Sn0.050MoO3、Sn0.063MoO3分別和1.4 g S混合,加入 25 mL無水乙醇混合3 h。將混合后的漿料置于在60℃真空干燥箱中干燥12 h,將干燥后的粉末轉移到管式爐中在Ar氣(99.999%)的氣氛條件下以1℃/min的升溫速率升高到155℃,保溫12 h,然后自然冷卻得到樣品。